En febrero de 2026 medios de todo el país se hicieron eco de una noticia sobre un avance en la detección precoz de la esclerosis lateral amiotrófica (ELA) mediante el análisis de células sanguíneas¹. La investigación, liderada por la Dra. Valle Palomo en IMDEA Nanociencia, fue publicada en el artículo científico “Agregación del proteoma en células derivadas de pacientes con Esclerosis Lateral Amiotrófica para la evaluación de fármacos personalizada”². El estudio propone utilizar linfocitos obtenidos de muestras de sangre para estudiar procesos celulares característicos de la enfermedad y probar tratamientos de forma personalizada. Sin embargo, como ocurre a menudo en ciencia, el hallazgo es prometedor, pero todavía está lejos de convertirse en una herramienta clínica. Comprender qué han descubierto exactamente los investigadores nos ayuda a situar este avance en su contexto real.
El contexto biológico: agregados anómalos de proteínas
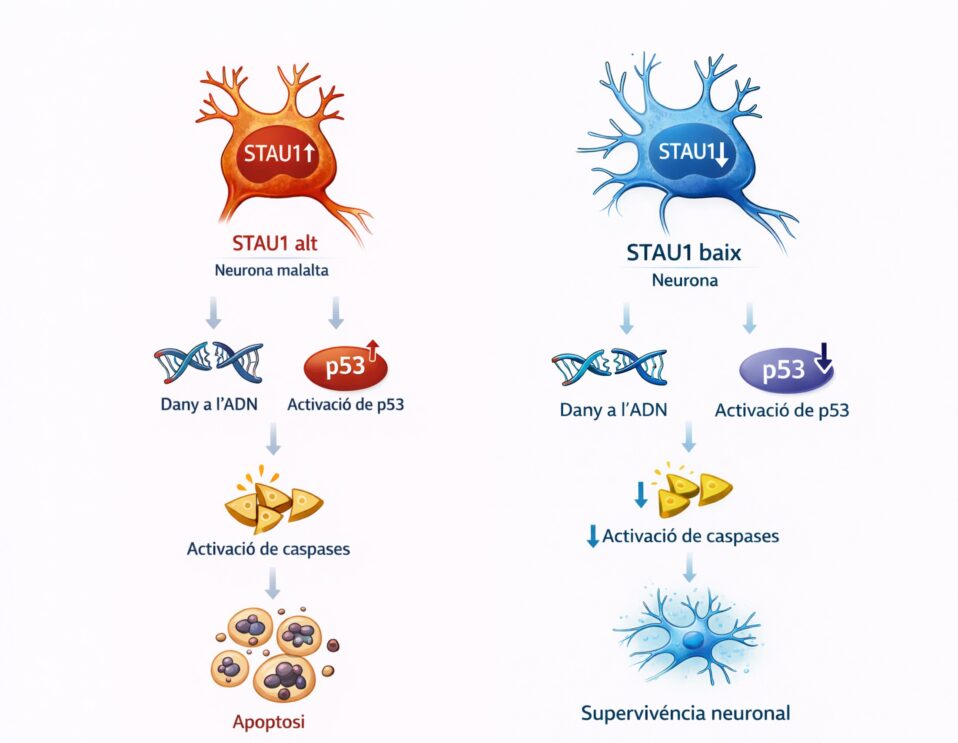
La ELA es una enfermedad neurodegenerativa en la que las motoneuronas, las células del sistema nervioso encargadas de controlar los músculos, van perdiendo capacidades y muriendo progresivamente. Uno de los mecanismos celulares más estudiados en esta enfermedad (y en otras enfermedades neurodegenerativas como el Alzheimer o el Parkinson) es la agregación anómala de proteínas. En el caso de la ELA, una de estas proteínas es la TDP-43³.
En células sanas, la TDP-43 se encuentra principalmente en el núcleo celular, donde participa en procesos esenciales como la regulación de la expresión génica. Sin embargo, en muchos pacientes con ELA, la proteína TDP-43 sufre una modificación anómala llamada hiperfosforilación. Esto significa que se añaden grupos de moléculas de fosfato en lugares indebidos de la proteína, lo que altera su estructura. Cuando esto ocurre, la proteína cambia de forma, pierde su función normal y tiende a unirse con otras copias mal plegadas, formando los llamados “agregados” fuera del núcleo, en el citoplasma de las motoneuronas. Estos agregados anómalos de proteína son tóxicos para la motoneurona y contribuyen al proceso de neurodegeneración³.
Un enfoque diferente: estudiar células sanguíneas
En lugar de estudiar neuronas directamente, ya que no se pueden obtener de pacientes vivos, el equipo de investigación exploró una alternativa más accesible: las células de la sangre. En la sangre circulan los linfocitos, células del sistema inmunitario que pueden aislarse en el laboratorio. Los investigadores tomaron linfocitos de pacientes con ELA y los sometieron a un proceso llamado inmortalización, que permite trabajar durante semanas o meses con células obtenidas directamente de los pacientes⁴.
Al analizar estas células, los investigadores observaron que los linfocitos inmortalizados provenientes de pacientes con ELA esporádica mostraban mayores niveles de agregados proteicos que los de individuos sanos, tal como sabemos que ocurre en las motoneuronas de los pacientes. Esto sugiere que algunas alteraciones celulares características de la enfermedad también podrían, en algunos casos, detectarse en células sanguíneas. Por lo tanto, los linfocitos podrían ser útiles como modelo de laboratorio, dado que las células sanguíneas son más accesibles y se pueden obtener fácilmente de los pacientes².
Un posible uso: probar tratamientos de forma personalizada
Una vez establecido este modelo celular, los investigadores exploraron una segunda posibilidad: evaluar la respuesta de las células de cada paciente a distintos fármacos. Para ello, probaron compuestos que ya habían mostrado potencial en otros estudios, como inhibidores (bloqueadores) de quinasas, que son las enzimas responsables de añadir grupos fosfato a proteínas como la TDP-43. En teoría, si el modelo en linfocitos actuara como se esperaba, bloquear estas enzimas podría evitar la hiperfosforilación de la proteína y reducir la formación de los agregados tóxicos mencionados anteriormente⁵.
Algunos de estos fármacos mostraron cierta reducción de la agregación proteica en linfocitos cuando se analizaban los resultados de forma global. Sin embargo, al estudiar las células de cada paciente por separado, la respuesta variaba mucho entre individuos². Este resultado refleja una realidad bien conocida en la ELA: la enfermedad es extremadamente heterogénea. Los pacientes pueden presentar diferentes causas genéticas, síntomas iniciales o velocidades de progresión. Precisamente por ello, herramientas como este modelo celular podrían ayudar en el futuro a evaluar qué tratamientos funcionan mejor en cada paciente, un paso importante hacia la medicina personalizada².
Es importante aclarar que el estudio no pretende buscar nuevos fármacos, sino generar un modelo celular de laboratorio accesible para el desarrollo de fármacos y, en el futuro, poder estudiar cómo cada paciente reaccionaría a las diferentes terapias disponibles.
Mirando al futuro
Aunque todavía queda mucho camino hasta que estas técnicas se traduzcan en aplicaciones clínicas, en ciencia los grandes cambios rara vez ocurren de un día para otro. Antes de poder probar fármacos en este sistema, los investigadores tuvieron que demostrar que las alteraciones proteicas observadas en las neuronas también aparecen en linfocitos, que estas células pueden mantenerse de forma estable en cultivo y que el modelo reproduce características relevantes de la enfermedad. Cada uno de estos pasos representa un pequeño avance. La noticia no anuncia una cura inmediata ni una nueva prueba diagnóstica disponible mañana, pero sí señala que la investigación está explorando nuevas formas de entender la ELA y de acercarse a un objetivo fundamental de la medicina moderna: detectar la enfermedad lo antes posible y desarrollar tratamientos adaptados a cada paciente.
Autors
Marta Vela Martínez¹, Pol Mengod Soler¹i Dr. Pol Andrés Benito²
1. Investigadora Predoctoral en el grupo de Neurogenética y Enfermedades Neurológicas del Centro de Investigación Biomédica de Bellvitge (IDIBELL).
2. Investigador Principal en el Grupo de Estudio de Cognición y Conducta del Instituto de Investigación Biomédica de Lleida (IRBLleida).

Referències
- ¿Se puede detectar la ELA de una manera precoz, rápida y poco invasiva? La Vanguardia
[Internet]. 2026 Feb 23 [citado 2026 Mar 18]. Disponible en:
https://www.lavanguardia.com/ciencia/20260223/11451382/detectar-ela-manera-precozrapida-poco-invasiva.html - Pérez de la Lastra Aranda C, Tosat-Bitrián C, Porras G, Dafinca R, Muñoz-Torrero D, Talbot K,
Martín-Requero Á, Martínez A, Palomo V. Proteome aggregation in cells derived from amyotrophic lateral sclerosis patients for personalized drug evaluation. ACS Chem Neurosci. 2024 Nov 6;15(21):3945-3953. DOI:10.1021/acschemneuro.4c00328 - Cohen TJ, Lee VM, Trojanowski JQ. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol Med. 2011 Nov;17(11):659-67. DOI:10.1016/j.molmed.2011.06.004
- Posa D, Martínez-González L, Bartolomé F, Nagaraj S, Porras G, Martínez A, Martín-Requero Á. Recapitulation of pathological TDP-43 features in immortalized lymphocytes from sporadic ALS patients. Mol Neurobiol. 2019 Apr;56(4):2424-2432. DOI: 10.1007/s12035-018-1249-8
- Alquézar C, Salado IG, de la Encarnación A, et al. Targeting TDP-43 phosphorylation by Casein Kinase-1δ inhibitors: a novel strategy for the treatment of frontotemporal dementia. Mol Neurodegener. 2016;11:36. DOI:10.1186/s13024-016-0102-7